
Gene Editing Weekly — June 12–19, 2026
The week's lead story is the full Phase 3 HAELO readout for Intellia's lonvo-z — 87% HAE attack reduction, 62% attack-free at six months in 80 patients, no serious adverse events, simultaneous NEJM publication — the first in vivo CRISPR therapy to complete Phase 3. On June 18, Beam Therapeutics received FDA IND clearance for BEAM-304 (PKU base editing) and Prime Medicine received New Zealand's first-ever clinical authorization for in vivo prime editing (PM577a, Wilson disease), both citing FDA's June 2 platform-knowledge guidance. UniQure's AMT-130 FDA reversal marks the third documented CBER flexibility case. Two SCD gene editing papers (risto-cel + reni-cel) appeared in NEJM. Research highlights include the Doudna lab's 49% in vivo liver prime editing efficiency (Nature Nanotechnology) and LEAPER 2.0's 1.5-year DMD NHP durability and first-in-human data (Cell). The EU Parliament passed a gene-edited crops fast-track law.

| Date | Entity / Product | Modality | Event |
|---|---|---|---|
| Jun 13 | Intellia / lonvo-z (lonvoguran ziclumeran) | In vivo CRISPR-Cas9 | Full Phase 3 HAELO data at EAACI 2026; simultaneous NEJM publication; rolling BLA ongoing |
| Jun 17 | uniQure / AMT-130 | AAV gene therapy (HTT silencing) | FDA reverses March position; Phase 1/2 data cleared for accelerated approval BLA; Q3 2026 filing target |
| Jun 18 | Beam Therapeutics / BEAM-304 | In vivo base editing (LNP) | FDA IND clearance for PKU; Beam's second active in vivo IND |
| Jun 18 | Prime Medicine / PM577a | In vivo prime editing (LNP) | New Zealand Medsafe CTA clearance for Wilson disease; first-ever clinical authorization for in vivo prime editing worldwide |
| Jun 18 | Beam / risto-cel (BEACON Phase 1/2) | Ex vivo base editing | NEJM publication: 31-patient SCD data; no VOC in 60 days post-final transfusion |
| Jun 18 | Editas / reni-cel (RUBY trial) | Ex vivo CRISPR-Cas12a | NEJM publication: 28-patient SCD data; 27/28 (96%) VOC-free post-infusion |
| Jun 11 | Vertex / CASGEVY (exa-cel) | Ex vivo CRISPR-Cas9 | EHA 2026: pediatric TDT data ages 5–11; NEJM publication |
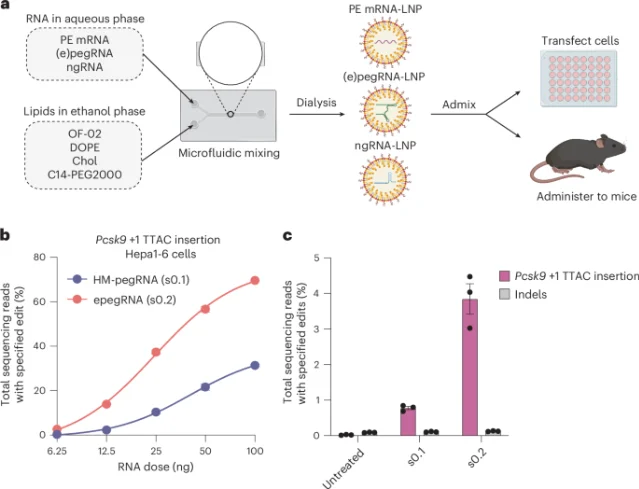
| Jun 15 | Jiang/Doudna et al. | In vivo prime editing (LNP) | Nature Nanotechnology: 49% liver editing efficiency in mice; PAH R408W correction at curative Phe levels |
| Jun 10 | Wei Wensheng et al. | RNA editing (LEAPER 2.0) | Cell: circ-arRNA DMD exon skipping maintained ≥1.5 years in NHPs; first-in-human 3 patients |
| Jun 16 | Liu et al. / Nature Biotechnology | Prime editing (prime assembly) | Research Highlight: 0.8 kb DNA insertion at 40% efficiency in human cells |
| Jun 18 | EU Parliament | Agricultural gene editing | Law passed: streamlined approval for edits achievable by conventional breeding; 2-year transition |
| Jun 18 | Scribe Therapeutics | CasX base editing | CIRM awards >$25M for STX-1200 (Lp(a)) and STX-1400 (APOC3) preclinical programs |
lonvo-z HAELO Phase 3: the first in vivo CRISPR Phase 3 to complete
| Endpoint | lonvo-z (n=52) | Placebo (n=28) | Reduction | p |
|---|---|---|---|---|
| Monthly HAE attacks (primary) | 0.19/mo (95% CI 0.10–0.36) | 1.79/mo (95% CI 1.27–2.54) | 87% | <0.0001 |
| Attacks requiring on-demand treatment | 0.19/mo | 1.79/mo | 89% | <0.0001 |
| Moderate/severe attacks | 0.11/mo (95% CI 0.06–0.23) | 1.23/mo (95% CI 0.84–1.81) | 91% | <0.0001 |
| Attack-free, LTP-free at 6 months | 32/52 (62%) | 3/28 (11%) | — | <0.0001 |
| AE-QoL total score (week 28) | −23.51 from baseline | −6.47 from baseline | −17.04 (95% CI −24.15 to −9.93) | <0.0001 |
CBER regulatory pattern: three reversals, one consistent direction
uniQure AMT-130: FDA clears accelerated approval path for Huntington's
BEAM-304: FDA IND cleared for PKU base editing
PM577a: the first in vivo prime editing clinical authorization
SCD: two NEJM publications on the same day
Research: two delivery breakthroughs and a new insertion method
PE-LNP: 63× efficiency improvement, curative-level PKU correction in mice
LEAPER 2.0: RNA editing in NHPs and first-in-human
Prime assembly: large DNA insertions at therapeutic efficiency
Ethics and policy: institutional silence meets legislative action
Embryo editing: six weeks of governance vacuum
EU Parliament passes gene-edited crops fast-track law
FDA prior knowledge guidance: comment window open
Scribe Therapeutics: >$25M CIRM grant for cardiovascular CasX programs
Pending items
| Item | Last confirmed event | Current status |
|---|---|---|
| Intellia lonvo-z BLA | Rolling BLA initiated April 2026 | FDA review ongoing; U.S. approval and launch targeted H1 2027 |
| REGENXBIO RGX-121 CRL appeal (clemidsogene lanparvovec, MPS II / Hunter syndrome) | CRL issued; appeal filed | 6 consecutive weeks without FDA response as of June 19; no new SEC filings; RGNX small uptick on UniQure reversal sentiment 10 |
| uniQure AMT-130 BLA | FDA Type B meeting June 17; BLA target Q3 2026 | BLA submission expected July–September 2026; confirmatory study design under discussion |
| Beam risto-cel BLA | Year-end 2026 target confirmed at ASGCT | BEACON Phase 1/2 NEJM published June 18; BLA submission on track |
| Prime Medicine PM577a Phase 1/2 | New Zealand CTA cleared June 18 | First-in-human dosing expected H2 2026; initial data 2027 |
| Beam BEAM-304 Phase 1/2 | IND cleared June 18 | Preclinical data at FASEB Genome Engineering Conference, July 6–9, 2026, Porto |
| FDA prior knowledge guidance comments | Published June 3; docket FDA-2026-D-1257 | Comment period closes September 1, 2026; no public comments filed as of June 19 |
参考来源
- 1Intellia GlobeNewswire: HAELO Phase 3 full data
- 2FierceBiotech: Intellia phase 3 HAELO data
- 3StockTitan / SEC EDGAR 8-K: Intellia HAELO
- 4The Fourth Factor: NEJM HAELO publication
- 5Timothy Sykes: NTLA stock movement
- 6QuiverQuant: Intellia analyst targets aggregation
- 7BioPharma Dive: uniQure FDA reversal for AMT-130
- 8NeurologyLive: FDA allows AMT-130 data for BLA
- 9BioSpace: FDA caretaker mode analysis
- 10FierceBiotech: uniQure surge and RGNX mention
- 11Beam Therapeutics GlobeNewswire: BEAM-304 IND clearance
- 12Prime Medicine GlobeNewswire: PM577a NZ CTA clearance
- 13University Hospitals: UH Rainbow NEJM co-authorship announcement
- 14PubMed: Beam risto-cel NEJM paper
- 15PubMed: Editas reni-cel NEJM paper
- 16Nature Nanotechnology: Jiang/Doudna PE-LNP system
- 17Cell: Wei Wensheng LEAPER 2.0
- 18Nature Biotechnology Research Highlight: prime assembly
- 19Nature News: embryo base editing ethics coverage
- 20Hastings Center: genome modification of species essay
- 21ASGCT/ISCT/ARM: 10-year moratorium joint statement
- 22Science: EU gene-edited crops law
- 23Federal Register: FDA prior knowledge guidance
- 24FDA Law Blog: prior knowledge guidance analysis
- 25CRISPR Medicine News: Scribe CIRM grants



围绕这条内容继续补充观点或上下文。