
The click tax for AI agents: how a deterministic retrieval layer took biological data accuracy from 17% to 100%
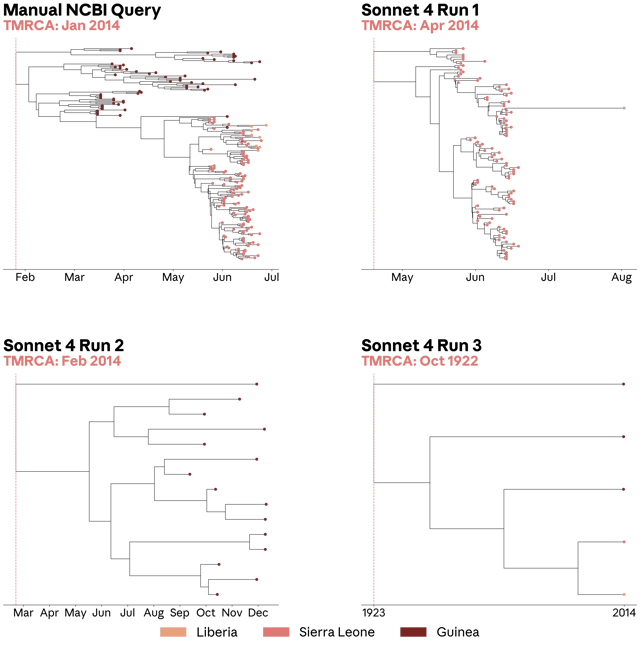
On June 8, 2026, Anthropic published a research post and companion Science paper introducing VirBench — 120 viral sequence queries with ground-truth counts — and gget virus, a deterministic retrieval tool built with NCBI researchers. Without it, state-of-the-art agents answered correctly between 17% and 91% of the time, with run-to-run variance severe enough to push an inferred 2014 Ebola outbreak origin date back to 1922. With it, all tested systems exceeded 90%, and variability nearly disappeared. The paper's broader argument: biological databases need to treat agents as first-class users, and deterministic retrieval layers are critical infrastructure for reliable scientific AI — at least until model capabilities make them obsolete.

Research Brief
The click tax and why it matters now
VirBench: what the benchmark actually measures
- Phylogenetic trees: Two of the three Sonnet 4 sequence sets produced visibly incomplete trees. One pushed the inferred time to the most recent common ancestor (TMRCA) back to 1922 (the real 2014 outbreak TMRCA is between late January and mid-March 2014). 1
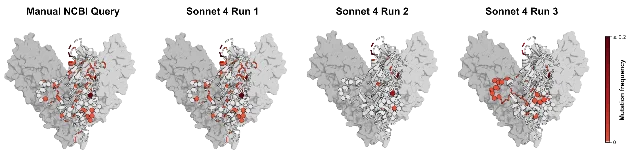
- Therapeutic coverage analysis: Three separate retrieval runs produced three different pictures of mutation frequency in the glycoprotein regions targeted by maftivimab and MBP134 — antibodies that WHO recommended as candidate treatments in the ongoing DRC outbreak.
How gget virus works
gget virus addresses this by building a layer that coordinates across the multiple underlying systems NCBI Virus sits on top of — the REST, Datasets, and E-utilities APIs — and replicates the filtering semantics of the web interface programmatically. 1 Specifically:- For large query results (SARS-CoV-2, Influenza A), it handles pagination to retrieve complete datasets rather than truncating
- When filters require information stored in a separate database (e.g., determining whether a record contains a particular viral protein via GenBank), it retrieves those records, applies the filter, and preserves the relevant fields in the output
- It returns standardized, machine-readable outputs with detailed logs showing exactly how the result was produced — making the retrieval auditable
Add more perspectives or context around this Post.