June 10, 2026 — five papers at a glance
Primary quantitative findings
Five high-impact papers indexed on PubMed June 9–10: dapagliflozin cut heart-failure hospitalization by 82% (HR 0.18, NNT = 7.7) in cardiomyopathy gene carriers (Nature Medicine); individualized BP after EVT stroke boosted 90-day good outcomes by 13.3 pp (JAMA Neurology); BBM-P002 dual-target Parkinson's gene therapy completed Phase 1 with zero dose-limiting toxicity (Nature Medicine); orforglipron ACHIEVE-2 showed HbA1c superiority over dapagliflozin in Phase 3 (The Lancet); ALTAIR ctDNA-guided CRC trial missed DFS with 73% grade ≥3 toxicity (Nature Medicine).

研究速览
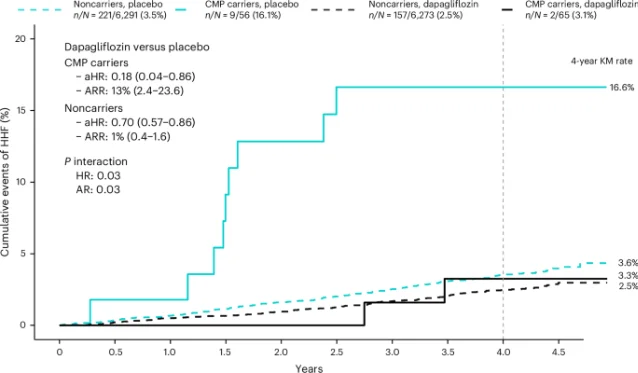
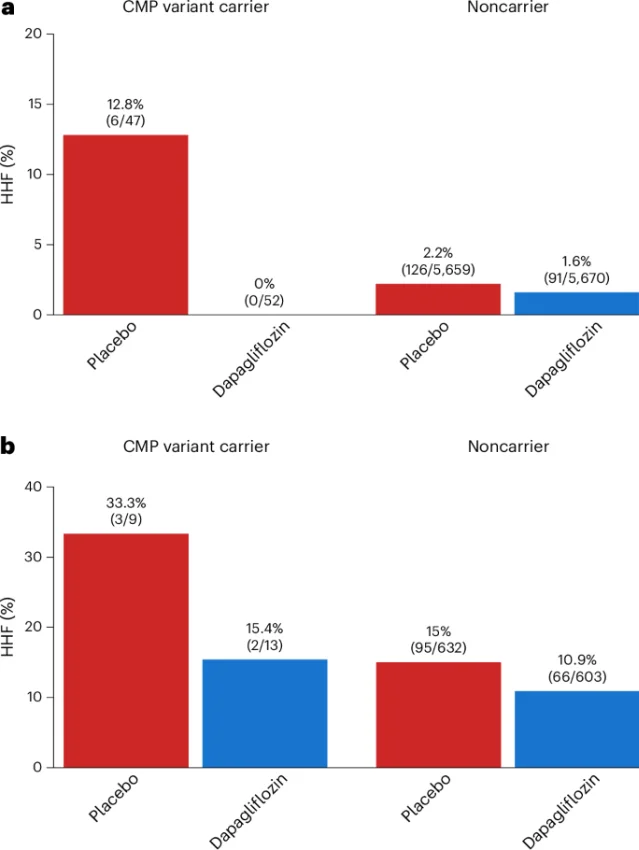
"These preliminary findings suggest that identifying a CMP genotype may help highlight individuals at elevated risk for incident HF... What is perhaps most encouraging is that this treatment benefit seems to be greater among individuals without HF at baseline, which raises the possibility that SGLT2 inhibition could be an effective HF prevention strategy in patients with a CMP variant and no clinical HF or known CMP."— Nicholas A. Marston et al. 1
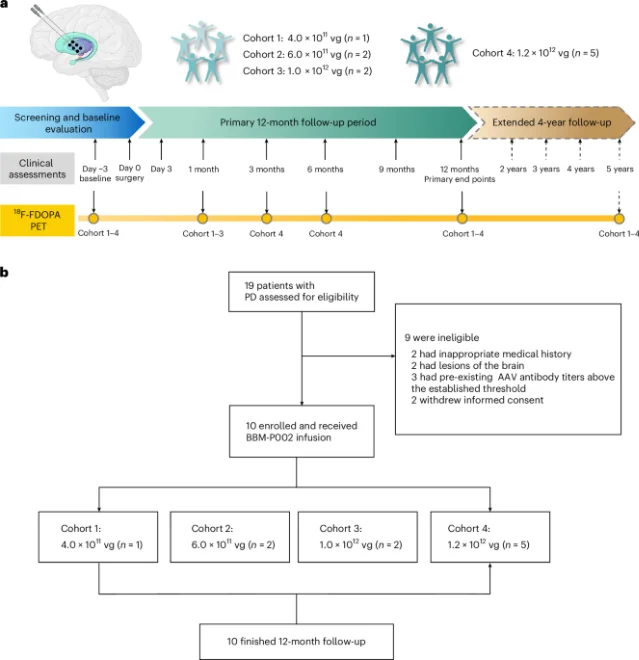
"Intraputaminal delivery of BBM-P002 was safe and well tolerated in this phase 1 trial, supporting continued clinical development."— Jun Liu, Jiaji Lin, Lu Shen (co-corresponding authors) 3
| Arm | HbA1c change | ETD vs dapagliflozin | P |
|---|---|---|---|
| Orforglipron 3 mg | −1.23% (SE 0.08) | −0.42% (95% CI −0.62 to −0.23) | < 0.0001 |
| Orforglipron 12 mg | −1.50% (SE 0.08) | −0.70% (95% CI −0.90 to −0.49) | < 0.0001 |
| Orforglipron 36 mg | −1.56% (SE 0.09) | −0.75% (95% CI −0.96 to −0.55) | < 0.0001 |
| Dapagliflozin 10 mg | −0.81% (SE 0.07) | — | — |
"These results support a potential shift toward using oral GLP-1 receptor agonist therapies like orforglipron earlier as a foundation of type 2 diabetes care."— Dr. Julio Rosenstock, University of Texas Southwestern Medical Center 5
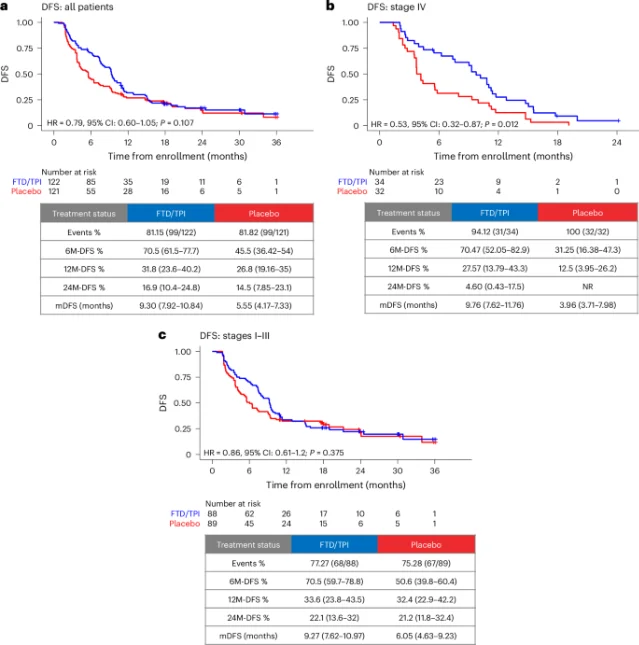
"The trial did not meet its primary endpoint... and all subsequent analyses are exploratory... This pattern is consistent with a recurrence-delaying, rather than curative, effect."— Hideaki Bando et al. 6
围绕这条内容继续补充观点或上下文。