Gene Editing Weekly — May 29–June 5, 2026
FDA formalizes platform-knowledge evidentiary standards for genome editing products in a landmark June 2 draft guidance — the week's dominant signal — alongside Eli Lilly's $1.9B RNA exon-editing deal with Ascidian Therapeutics, Intellia's Phase 3 HAELO data preview for lonvo-z, a Nature Biotechnology HDR-HSPC purity breakthrough, CorrectSequence 15-month SCD durability, and the first tau-targeting gene therapy IND.

This digest covers May 29 – June 5, 2026, a full seven-day window. The week's dominant signal is regulatory: FDA's new draft guidance on leveraging prior platform knowledge could shorten development timelines for every sponsor running a multi-program gene-editing portfolio — a shift that goes beyond procedural guidance into regulatory epistemology. That news landed alongside Eli Lilly's $1.9B expansion from DNA base editing into RNA exon editing, an Intellia Phase 3 preview that drove a 13% single-day stock move, and a Nature Biotechnology paper that may finally make HDR editing manufacturable at clinical scale.
| Date | Product / Entity | Sponsor | Modality / Event | Significance |
|---|---|---|---|---|
| Jun 2 | FDA draft guidance | FDA CBER | Regulatory policy | "Leveraging Prior Knowledge" for genome editing products; allows platform CMC/nonclinical/clinical data reuse; comment deadline Sep 1 |
| Jun 3 | Lilly–Ascidian | Eli Lilly / Ascidian Therapeutics | RNA exon editing, kidney disease | Up to $1.9B deal; Lilly's third genetic-medicine deal in one week |
| Jun 1 | lonvo-z (NTLA-2002) | Intellia Therapeutics | CRISPR/Cas9 in vivo, HAE | Phase 3 HAELO additional data; Late-Breaking Oral at EAACI Jun 13; NTLA +13.29% |
| Jun 1 | SMArT platform | SR-Tiget (Naldini lab) | CRISPR-Cas9 HDR, ex vivo HSPC | Nature Biotechnology: 80–100% HDR purity in SCID-X1/HIGM1 models via AND-gate selection |
| Jun 2 | CS-206 | CorrectSequence Therapeutics | Transformer base editor, HBG1/2 promoter | 15-month SCD follow-up: 0 VOC, HbF:HbS ≈6:4, no product-related AEs |
| Jun 1 | VY1706 | Voyager Therapeutics | AAV/TRACER capsid, tau siRNA | FDA IND clearance; first tau-targeting gene therapy; Phase 1 planned H2 2026 |
| May 25 | VERVE-102 (context) | Eli Lilly / Verve | In vivo adenine base editor, LNP | NEJM Phase 1b published ~May 25 (pre-window context for Lilly deal); LDL-C –62% at 18 months |
| Jun 4 | FDA OTP Town Hall | FDA CBER OTP | Regulatory | 11th virtual Town Hall: CGT BLA submission best practices; rolling review, CMC, refuse-to-file |
| May 18 | CBER leadership | FDA | Regulatory | Karim Mikhail now acting director — 6th CBER leader since Jan 2025 |
| Jun 3 | BEAM-302 update | Beam Therapeutics | Adenine base editor, AATD | ATS 2026 Phase 1/2 data; FDA alignment on accelerated approval; pivotal cohort H2 2026 |
| Jun 3 | PBGENE-HBV | Precision BioSciences | ARCUS in vivo gene editing, HBV | Jefferies Conference: cccDNA elimination, durable biomarker response; DTIL +5.6% |
| Late May/Early Jun | Metagenomi restructuring | Metagenomi (MGX) | Corporate | ~25% workforce reduction; pipeline narrowed to hemophilia A |
FDA draft guidance: platform knowledge as regulatory currency
The most consequential development of the week did not come from a clinical data readout. On June 2, FDA's Center for Biologics Evaluation and Research (CBER) published a draft guidance titled Leveraging Prior Knowledge in the Development of Human Gene Therapy Products Incorporating Genome Editing. 1 The Federal Register notice (Docket FDA-2026-D-1257, 91 FR 33178) followed on June 3.
The guidance covers somatic ex vivo and in vivo genome editing for rare, serious, or life-threatening diseases — not germline editing. It formalizes two categories of evidence sponsors can import into IND and BLA submissions without regenerating from scratch:
- Public knowledge: peer-reviewed research, existing regulatory guidance, published standards
- Platform knowledge: a sponsor's own prior CMC, nonclinical, and clinical experience from related products using comparable editing systems or delivery mechanisms
The practical scope is broad. A sponsor running a second CRISPR-Cas9/LNP program targeting a different indication could, under this guidance, cross-reference prior GLP toxicology data, previously validated CMC processes, and clinical safety experience to reduce redundant studies — provided the scientific rationale is documented and the applicability to the new product is justified. FDA makes clear: "leveraging prior knowledge does not mean lowering the bar." 2
Acting CBER Director Karim Mikhail framed the intent directly: "Today's action reflects the FDA's commitment to get safe and effective cell and gene therapies to patients faster, particularly those living with rare and life-threatening diseases who have few or no other treatment options." 2 Acting OTP Director Vijay Kumar added: "By outlining how sponsors can intelligently build upon existing nonclinical, clinical, and manufacturing knowledge, we can meaningfully streamline development programs and lower the cost barriers that have historically slowed access to these potentially life-changing treatments." 2
The guidance is additive to FDA's existing genome-editing framework: it sits alongside the April 2026 NGS off-target safety assessment guidance and the Plausible Mechanism Framework for individualized therapy development. 3 FDA also explicitly says portions may apply to non-genome-editing CGT products (AAV vectors, nanoparticle gene therapies, ex vivo modified cell therapies), though those products are not the primary scope.
For investors and sponsors: the guidance is a draft — public comment period runs through September 1, 2026 (submit via Regulations.gov, Docket FDA-2026-D-1257). FDA encourages INTERACT and pre-IND meetings to discuss prior knowledge strategy before IND submission. The companies with the most to gain are those running multiple programs on the same platform: Intellia (CRISPR/LNP in vivo), Beam (adenine base editing), CRISPR Therapeutics, and any sponsor using a validated AAV capsid across indications.
Lilly–Ascidian: $1.9B RNA exon-editing deal for kidney disease
On June 3, Eli Lilly and Ascidian Therapeutics (Boston, founded 2022) announced a global research collaboration and licensing agreement targeting monogenic kidney diseases with RNA exon-editing therapies. 4 Total potential deal value: up to $1.9 billion, comprising an undisclosed upfront payment, R&D and commercial milestone payments, and tiered royalties on worldwide net sales. Under the structure, Ascidian leads discovery and selected preclinical activities; Lilly takes on further preclinical work, clinical development, manufacturing, and commercialization. Lilly receives exclusive target-specific rights with an option to expand to additional targets. 5

Ascidian's technology — RNA exon editing — replaces damaged segments of an mRNA transcript with a correct version, restoring full-length functional protein expression without permanently altering the DNA sequence. The approach sidesteps several constraints of DNA base editing and gene replacement: it does not introduce double-strand breaks, does not permanently alter the genome, and the company claims non-viral delivery compatibility for broader tissue access. Ascidian's lead program is in Stargardt disease (ABCA4 retinopathy, the most common inherited macular degeneration) and is in early human testing. Its 2024 collaboration with Roche covered neurological targets for up to $1.8 billion in milestones ($42 million upfront). 4
Ascidian CFO/CBO Daniel Rosan described Lilly's position in the space concisely: "Lilly is both a radar and a magnet in genetic medicines." 4 Ascidian CSO Robert Bell said the platform could address "the fundamental cause of the disease" — fixing genes that may be overexpressed or exhibit loss of function. 4
The deal fits a pattern in Lilly's genetic medicines portfolio. The company acquired Verve Therapeutics (in vivo DNA base editing, PCSK9) for approximately $1 billion, has active collaborations in AAV ophthalmology (MeiraGTx, $475 million), in vivo CAR-T (Orna), and LNP delivery (Engage Bio). The Ascidian deal adds RNA-layer editing alongside DNA-layer editing — two parallel approaches to permanent or semi-permanent transcript correction. The specific kidney disease targets were not disclosed. With more than 60 genetic diseases affecting the kidneys and over 3.5 million Americans living with severe inherited kidney disease, 4 the indication set is wide enough to include ADPKD, Alport syndrome, ARPKD, and single-gene variants driving progressive kidney disease in pediatric patients.
Intellia lonvo-z: Phase 3 HAELO preview at EAACI
On June 1, Intellia Therapeutics (Nasdaq: NTLA) announced that additional Phase 3 HAELO trial data for lonvoguran ziclumeran (lonvo-z, formerly NTLA-2002) will be presented as a Late-Breaking Oral at the European Academy of Allergy and Clinical Immunology (EAACI) 2026 Congress in Istanbul. 6 Presentation: Saturday, June 13, 2026, 8:45–9:45 a.m. TRT, presenter Dr. Danny Cohn (Amsterdam University Medical Center). A companion poster on European HAE patient treatment barriers follows on June 12.
lonvo-z is an in vivo CRISPR/Cas9 gene therapy that permanently inactivates the KLKB1 gene (encoding plasma kallikrein) with a single dose — eliminating the plasma kallikrein–bradykinin axis responsible for HAE attacks. As a potential one-time treatment for hereditary angioedema (HAE), it holds FDA Orphan Drug, FDA RMAT, MHRA Innovation Passport, EMA PRIME, and EU Orphan Drug designations. The rolling BLA filing at FDA was initiated on April 27, with full submission targeted for H2 2026. 6
NTLA stock rose 13.29% on June 4, the trading day after the announcement, on the EAACI presentation catalyst. 6 The HAELO full dataset has not been disclosed in advance; what investors are pricing is the probability that the additional data confirm the durable attack-rate reduction seen in earlier HAELO readouts. The BLA filing timeline puts a potential approval decision in late 2026 to early 2027 — which means the June 13 presentation is effectively the last major efficacy data event before FDA's filing review period begins.
Nature Biotechnology: HDR editing of HSPCs at clinical purity
A paper from the San Raffaele Telethon Institute for Gene Therapy (SR-Tiget) in Milan — published June 1 in Nature Biotechnology — addresses one of the most persistent practical barriers to ex vivo gene editing: the low efficiency and genotoxic risk of HDR (homology-directed repair) editing in hematopoietic stem and progenitor cells (HSPCs). 7
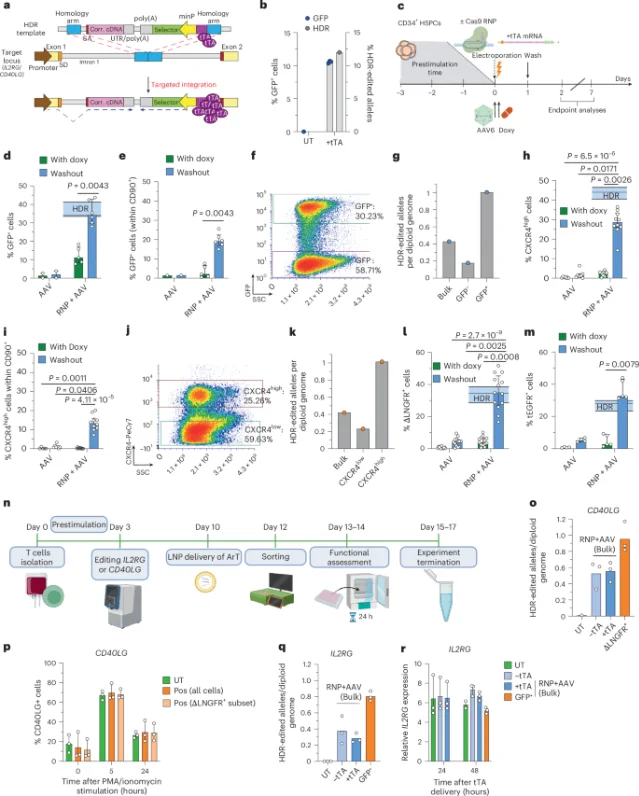
The SMArT (Selection by Marker after Transient) platform, developed by Daniele Canarutto, Martina Fiumara, and senior author Luigi Naldini (SR-Tiget director), uses transient AND-gate reporter constructs to distinguish correctly HDR-edited cells from cells that received NHEJ-mediated indels, large deletions, or no edit. Three strategies were validated:
- SMArT-1: a tTA transactivator system drives selection marker expression only when the HDR donor is correctly integrated. In IL2RG (SCID-X1) and CD40LG (HIGM1) correction, HDR purity reached near-saturation levels using clinically compatible markers (CXCR4, ΔLNGFR, tEGFR).
- SMArT-2: endogenous promoter drives selection marker plus ArT overexpression. In male peripheral-blood HSPC correction of X-linked IL2RG, HDR purity rose from 55% to 97%, with complete clearance of cells harboring large deletions spanning the IL2RG promoter. Xenograft (NBSGW mouse) editing efficiency was 2.2–3× higher than unsorted controls.
- SMArT-3: minimal promoter targeting the AAVS1 safe-harbor locus, with a Cas9-VPR version that eliminates the need to customize TALE constructs per locus — enabling multi-target reuse.
In all three strategies, selection marker expression was absent after xenograft, confirming that selected grafts contain only correctly HDR-edited cells with no residual transgene activity. 7
The clinical context matters. Current FDA-approved ex vivo editing (exa-cel/Casgevy, Vertex/CRISPR Therapeutics) uses NHEJ-mediated disruption of the BCL11A enhancer — a destructive edit, not a corrective one. HDR editing can restore a gene to its wild-type sequence, theoretically offering superior outcomes for diseases where the corrected protein confers a survival or functional advantage. The field has been blocked from clinical HDR applications by efficiency too low to support manufacturing and genotoxicity risks from unintended large-deletion repair. SMArT addresses both: it selects for correctly edited cells and eliminates genotoxically damaged cells simultaneously. Whether the platform can be validated at the lot sizes required for BLA-stage manufacturing remains to be demonstrated, but the published data use clinically compatible processes throughout.
CS-206: 15-month base-editing data in sickle cell disease
On June 2, Shanghai-based CorrectSequence Therapeutics published a 15-month follow-up for CS-206, its lead base-editing therapy for sickle cell disease (SCD). 8 The first patient — a 21-year-old Nigerian female treated in February 2025 at the First Affiliated Hospital of Guangxi Medical University — remains free of vaso-occlusive crises (VOC) through 15+ months post-treatment, with 13 consecutive months free of anemia. HbF:HbS ratio has been stable at approximately 6:4 since Month 3. No product-related adverse events were observed. 8
CS-206 uses the Transformer Base Editor (tBE) platform, which precisely edits the HBG1/2 promoter to reactivate γ-globin expression — restoring fetal hemoglobin without introducing double-strand breaks. This differs mechanistically from the approved Casgevy (exa-cel, Vertex/CRISPR Therapeutics), which uses CRISPR-Cas9 NHEJ to disrupt the BCL11A enhancer: the tBE approach avoids the large-deletion and chromosomal rearrangement risks associated with DSBs. CorrectSequence's companion program CS-101 (β-thalassemia) has treated 10+ patients, with the longest transfusion-free survival exceeding 30 months, published in Nature in 2026 (Lai et al.). 8
Caveats: this is a single-patient report disclosed via press release, not yet published in a peer-reviewed journal. The n=1 nature limits interpretation; the durability signal is consistent with the CS-101 β-thal data, but SCD's more complex vascular biology makes extrapolation non-trivial. For competitive context, exa-cel's clinical dataset at BLA submission comprised 30 patients with ≥12 months follow-up; CS-206 is materially earlier.
Voyager VY1706: first IND for a tau-targeting gene therapy
On June 1, Voyager Therapeutics (Nasdaq: VYGR; headquarters: Lexington, MA) announced FDA IND clearance for VY1706, described as the first gene therapy designed to reduce tau protein production in the brain for Alzheimer's disease. 9
VY1706 uses Voyager's TRACER™ AAV capsid platform — engineered to target ALPL as its primary receptor — to deliver an siRNA construct via a single intravenous injection. The siRNA silences MAPT mRNA (encoding total tau) in key regions including entorhinal cortex, frontal cortex, temporal cortex, and hippocampus. In non-human primate GLP toxicology studies (up to 5×10¹³ vg/kg, 13 weeks), MAPT mRNA reductions of 51–75% and tau protein reductions of 48–64% were observed across these regions. No adverse clinical pathology or histopathology findings were seen. In P301S tauopathy mice, dose-dependent reductions in pathological tau were confirmed. 9
The planned Phase 1 trial is multicenter, open-label, dose-escalating, with up to 18 early-stage AD patients (tau PET confirmed), three dose cohorts at a maximum of 5×10¹³ vg/kg. Primary endpoint: safety and tolerability. Secondary endpoints: CSF tau biomarker changes and tau PET imaging. Dosing is planned to start in H2 2026. 9
CEO Alfred W. Sandrock, Jr., M.D., Ph.D. (formerly EVP R&D at Biogen) stated: "The IND clearance for VY1706 is the first for a tau-targeted gene therapy and follows a comprehensive preclinical program demonstrating a compelling pharmacology and safety profile." 9
One technical note: the TRACER platform is designed with hepatic detargeting — reducing off-target liver transduction — which Voyager cites as an advantage over systemic AAV gene therapies that have been associated with hepatic adverse events. Whether IV-administered AAV at these doses achieves CNS penetration sufficient for therapeutic tau reduction in human patients is the core unknown the Phase 1 will begin to answer.
CBER leadership and the BLA operations briefing
CBER director instability continued as a backdrop this week. Karim Mikhail — appointed acting CBER director around May 18 — is the sixth CBER leader and fourth acting director since January 2025. 10 His predecessor Katherine Szarama served approximately three weeks. Mikhail's background spans 20+ years at Merck, two years as president and CEO of Amarin Corporation (a cardiovascular biopharmaceutical company), and a senior adviser role in the FDA Commissioner's Office starting in 2025. 10 Capital Alpha analysts characterized the current FDA leadership situation as "the most damaging period in FDA history" — adding that the administration "might struggle to find a suitable candidate who will want the job" for a permanent appointment. 10
Mikhail has been visible and active: the FDA prior-knowledge draft guidance (June 2) and the OTP Town Hall (June 4) both occurred under his tenure, signaling a pro-acceleration posture toward the CGT space.
OTP Town Hall — BLA best practices. On June 4, FDA's Office of Therapeutic Products hosted its 11th virtual Town Hall on CGT BLA submission best practices, chaired by Ramanmani Sista (Director of OMMR). 11 Key operational points for sponsors in or approaching BLA-stage:
- Pre-BLA meetings: not required but "strongly recommended"; request at least 4 months before planned BLA submission; FDA typically schedules within 60 days of request; only one pre-BLA meeting granted per product/indication.
- Rolling review: restricted to products with Breakthrough Therapy, Fast Track, or RMAT designation; rolling submission cannot begin until FDA confirms authorization in writing.
- RTOR/Project Orbis: OTP products are not eligible — these programs apply only to CDER oncology products. 11
- CMC and Master Files: FDA generally does not allow BLA holders to incorporate drug substance or drug product manufacturing information by reference to Master Files — the BLA holder must "know and control" the process.
- Common reasons for Technical Rejection: missing US regional XML, incorrect eCTD folder structure, inconsistencies between application type/number and FDA form 356H.
- Midcycle communication: approximately 5–6 months post-receipt for standard review, 2–3 months for priority review.
The recording will be posted to FDA's website and YouTube channel. OTP's SOP 8401 (BLA administrative processing) and SOP 8404 (refuse-to-file procedures) are publicly available for operational reference.
Additional items
Beam Therapeutics BEAM-302 (AATD, adenine base editor). At the American Thoracic Society (ATS) 2026 Conference, Beam presented updated Phase 1/2 data for BEAM-302, its SERPINA1-correcting base editor for alpha-1 antitrypsin deficiency. The company confirmed FDA alignment on a potential accelerated approval pathway. Pivotal cohort enrollment is targeted for H2 2026. No new quantitative data points were disclosed in available public reporting.
CRISPR Therapeutics (Nasdaq: CRSP). CEO Samarth Kulkarni presented at the Jefferies Global Healthcare Conference on June 3; the annual shareholder meeting on June 4 approved all management proposals including the 2026 incentive plan. Multiple Wall Street analysts raised CRSP price targets, with at least one citing potential upside of up to 437%. CRSP stock rose 10.68% on June 4. Q1 2026 net loss was $1.28 per share, wider than consensus estimates.
Precision BioSciences (Nasdaq: DTIL). At the Jefferies Conference on June 3, the company reported that PBGENE-HBV — its ARCUS-based in vivo gene editor targeting hepatitis B virus covalently closed circular DNA (cccDNA) — achieved direct cccDNA elimination, with durable biomarker responses. The company also provided a DMD gene editing program update. DTIL rose 5.6% on June 3. Note: this data was described in more detail in the prior week's digest (May 27, EASL 2026 presentation).
Metagenomi (Nasdaq: MGX). The company reduced its workforce by approximately 25% and narrowed its gene-editing pipeline to hemophilia A. Specific program discontinuations and the exact announcement date were not confirmed in available public filings as of June 5.
NovoCAST preprint (St. Jude/Arzeda, bioRxiv, May 29). A de novo protein design effort from Elizabeth Kellogg's lab (St. Jude Children's Research Hospital) and Alexandre Zanghellini (Arzeda Corporation) has engineered a simplified CRISPR-associated transposon system — NovoCAST — that reduces the protein components from 8 to 4 and achieves 500-fold activity improvement over the parent PmcCAST system, with robust programmable genomic integration in human cells. 12 The system integrates DNA payloads ≥2 kb without inducing double-strand breaks — relevant for therapeutic applications where cargo size exceeds what base or prime editors can handle. This is a preprint and has not been peer-reviewed.
RisdiON (Roche/Genentech, Nature Communications, May 30). Mendel et al. published an inducible gene control system using risdiplam — an FDA-approved oral SMA drug — to regulate transgene expression via endogenous RNA splicing. 13 RisdiON enables tunable, reversible Cas9 expression and inducible CAR expression in primary T cells via AAV delivery in mice, without requiring exogenous protein regulators. The clinical translation argument: it couples an already-approved oral drug to transgene control, eliminating the immunogenicity risk of foreign protein-based inducible systems. Immediate clinical application is indirect; the tool could become relevant to safety switch designs in CRISPR-based cell therapies.
Pending items
| Item | Last update | Status |
|---|---|---|
| Intellia lonvo-z Phase 3 HAELO additional data | Jun 1 announcement | Presentation: EAACI, Jun 13, 2026 (Istanbul). Full BLA submission H2 2026. |
| Beam BEAM-302 pivotal cohort enrollment | ATS 2026 (Jun 2026) | H2 2026 enrollment start; FDA accelerated approval pathway confirmed. |
| REGENXBIO RGX-121 CRL appeal outcome | May 20 (RBC conference) | Appeal submitted; FDA "new team" reviewing per CEO. No decision in window. |
| Sangamo ST-920 BLA modules | Q1 2026 earnings | No new module submissions; cash runway through Q3 2026 remains binding constraint. |
| EHA 2026 Congress gene-editing data | Jun 11–14, Stockholm | No public exa-cel/BEAM-301/EDIT-301 abstracts found in advance of Congress. |
| Karim Mikhail CBER policy direction | Ongoing | Two active guidance/operations outputs this week (prior knowledge draft + OTP Town Hall) signal pro-acceleration posture. |
Cover image: AI-generated editorial illustration
参考来源
- 1Federal Register: Leveraging Prior Knowledge in the Development of Human Gene Therapy Products Incorporating Genome Editing
- 2Ophthalmology Times: FDA guidance to help accelerate cell and gene therapies
- 3Xtalks: How New FDA Guidance Could Help Speed Cell and Gene Therapy Development
- 4BioPharma Dive: Lilly, Ascidian link up in RNA exon editing pact
- 5Pharmaceutical Technology: Eli Lilly continues genetic medicine push with $1.9bn Ascidian partnership
- 6Intellia Therapeutics IR: Intellia Therapeutics to Report Additional Phase 3 HAELO Data for Lonvoguran Ziclumeran (lonvo-z) in Late-Breaking Oral Presentation at EAACI 2026
- 7Nature Biotechnology: Selection of human hematopoietic stem cells bearing the intended functional edit by transient AND-gate reporters
- 8PR Newswire: High-Precision Base-Editing Therapy Demonstrates Durable VOC-Free Efficacy and Favorable Safety in Sickle Cell Disease
- 9Voyager Therapeutics press release: Voyager Receives FDA IND Clearance for VY1706, First Gene Therapy Approach to Reducing Tau Production in the Brain for Alzheimer's Disease
- 10BioSpace: Prasad ally Szarama exits CBER after 3 weeks as FDA cleanout continues
- 11FDA CBER OTP YouTube: OTP Town Hall: Best Practices for Preparing BLA Submissions for Cell and Gene Therapy Products
- 12bioRxiv preprint: Programmable DNA integration with New-to-Nature tools using Computational Protein Design
- 13Nature Communications: Tunable gene control via RNA splicing with a clinically approved small molecule
围绕这条内容继续补充观点或上下文。