Claude vs. ChemDraw: Anthropic's NMR benchmark shows a general-purpose model matching dedicated chemistry software
On June 5, 2026, Anthropic published a white paper showing Claude Opus 4.7 matches or outperforms ChemDraw and MestReNova on NMR forward prediction — and can do structure elucidation from 1D spectra alone, a task the dedicated software leaves entirely to the chemist. The article covers the benchmark setup, the key accuracy numbers, the stated limitations, and what this means for Anthropic's broader AI-for-science program.

Vistazo a la investigación
A general-purpose model walks into a chemistry lab
NMR spectroscopy is not the kind of problem you'd expect a language model to be good at. It involves reading instrument readouts — peaks and splittings on a frequency axis — and either predicting what a molecule should look like in a magnetic field, or working backward from those peaks to figure out the structure of an unknown compound. Dedicated software like ChemDraw and MestReNova has been the standard for decades. They run deterministic algorithms tuned on curated spectral databases. They return the same answer every time.
On June 5, 2026, Anthropic published a white paper showing that Claude Opus 4.7 — a general-purpose model with no chemistry-specific fine-tuning — now matches or outperforms both tools on the forward prediction task, and can handle the inverse task (structure elucidation from spectra alone) that the dedicated software leaves entirely to the human chemist. 1
The results are more precise than typical "AI beats software" benchmarks, and the limitations are stated clearly enough to take seriously.
The two tasks
Forward prediction is the standard task: given the molecular structure of a compound (input as a SMILES string), predict the ¹H and ¹³C NMR spectrum — which peaks appear at which positions along the ppm axis, and what shape each peak takes. ChemDraw and MestReNova both do this.
Inverse prediction (structure elucidation) is harder: starting from an experimentally measured spectrum, plus the exact molecular formula from a high-resolution mass spectrometer, propose the structure of the unknown compound. This is where a chemist earns their pay. No general-purpose software does this from 1D NMR alone — it normally requires either 2D NMR experiments (COSY, HSQC, HMBC) or years of pattern-recognition experience.
Anthropic tested three Claude models (Opus 4.7, Opus 4.6, Sonnet 4.6) against ChemDraw and MestReNova on 20 novel compounds drawn from ChemRxiv preprints published after the models' training cutoff — a clean separation preventing any possibility of memorization. 1 The 20 compounds span four structural families, chosen to probe four distinct classes of NMR challenge.
The benchmark numbers
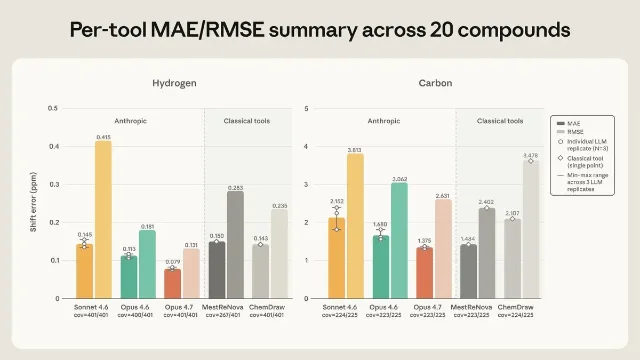
On hydrogen (¹H) forward prediction, Opus 4.7 achieved a mean absolute error of ±0.079 ppm — under half the ±0.20 ppm tolerance window a chemist considers correct. ChemDraw and MestReNova fell within that window less often. On carbon (¹³C), Opus 4.7 and MestReNova essentially tied, at ±1.37 and ±1.48 ppm respectively against a ±1.0 ppm tolerance.
Cargando gráfico…
The spread between models is telling. Sonnet 4.6 placed a notoriously mobile NH proton in the 10–13 ppm range when it actually appears around 6.8–7.9 ppm. Opus 4.6 scattered its guesses across several ppm. Opus 4.7 placed it slightly low but consistently so. This kind of difficult proton — a slow-exchange NH on an aminopyridazine scaffold in DMSO solvent — is the sort of thing that separates junior chemists from experienced ones.
Where the classical tools fell further behind was in peak shape prediction: Opus 4.7 matched the experimentally reported splitting pattern more often than any other tool, and all three Claude models predicted sub-peak spacing to within half a hertz about 80% of the time, against 26–35% for ChemDraw and MestReNova. 1 Splitting patterns are structurally informative — they encode how many neighboring protons a given hydrogen "sees" — so this matters beyond the headline accuracy numbers.
Structure elucidation: the task the software doesn't do
For the inverse task, Anthropic gave Opus 4.7 fifteen elucidation problems and asked it, three times each, to propose up to three ranked candidate structures. Each problem supplied the molecular formula (from HRMS) and the 1D ¹H and ¹³C spectra, without 2D data.
The fifteen were split by difficulty: eight simpler single-ring or two-fragment targets (formula + spectra only), and seven denser targets — fused rings, spirocycles — accompanied by the starting material's structure as an additional hint.
Opus 4.7 recovered all eight simpler structures on every attempt. On the seven harder targets with the starting-material hint, it returned the correct structure on all three attempts for four of them, and on two of three for the remaining three. 1
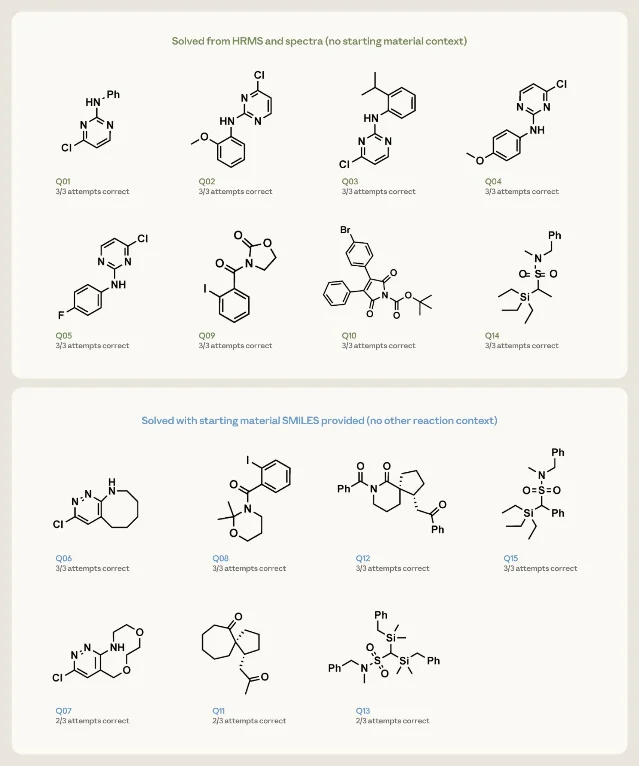
Dedicated structure-elucidation software has existed for decades, but it typically requires 2D NMR, specialized training, and licensed tools. Claude does this from the same 1D peak list a chemist would paste into a chat.
Stated limitations
The paper is notably honest about what these results don't show.
The evaluation covered 20 compounds for forward prediction and 15 for inverse. That's sufficient to establish rough performance rankings but not enough to characterize per-scaffold variance with statistical precision. The four structural families tested represent specific NMR challenges, not a random sample of small-molecule space.
Several categories were excluded by design: stereochemistry (1D NMR alone cannot fix configuration), natural product-scale complexity, 2D NMR experiments, and solvents beyond DMSO-d₆, CDCl₃, and D₂O. For the seven harder inverse targets, the model struggled without the starting-material hint — it would loop through reasoning without committing to a structure.
The authors also note that on denser inverse targets without starting-material context, the model sometimes failed to converge, suggesting that 1D NMR alone remains insufficient for highly complex unknowns regardless of model capability.
What's different about how Claude handles this
Classical NMR prediction tools work from curated databases of chemical shift increments — essentially lookup tables with additive correction rules. They don't generalize well to unusual environments or edge-case substituents, and they have no access to the reasoning process that a trained chemist would apply.
Claude approaches these problems differently. The paper shows, but doesn't explain fully, that the model appears to be doing something closer to chemical reasoning — it can articulate why a particular proton appears where it does, and show its work step by step. That auditability is one of the things the authors emphasize: a chemist can inspect the reasoning, not just accept or reject the output number.
The comparison with the inverse task is instructive. ChemDraw and MestReNova leave structure elucidation to the human entirely, because there's no algorithmic path from a 1D spectrum to a unique molecular structure without additional data or chemical judgment. The fact that Opus 4.7 can propose ranked candidates from the same 1D data a chemist would have suggests the model is applying something like the pattern-recognition reasoning that distinguishes expert chemists — at least for the classes of compounds tested.
The broader program
This white paper is the first output of a deliberate effort: Anthropic is working with synthetic, computational, and analytical chemists to extend Claude's chemical capabilities across a set of bottlenecks the authors identify explicitly. 1 The roadmap they describe includes:
- Reading and rendering chemical structures (converting drawings, patents, figures into machine-readable form)
- Reaction and synthetic reasoning (proposing routes, predicting outcomes, thinking through selectivity)
- Mechanism (explaining reactions in the electron-arrow language chemists use)
- Literature understanding (parsing the way compounds appear across publications — drawings, names, abbreviations, registry codes)
NMR prediction is the furthest along and the most benchmarkable. Retrosynthesis planning, the team notes, is "still being scoped." They're also expanding the AI for Science program specifically to chemistry research, inviting chemists working on problems where this kind of multimodal reasoning might help.
The comparison to similar "Claude as scientist" work is clarifying context: on the same day, Anthropic published a companion piece on deploying agents in biology — specifically a benchmark showing that AI agents given access to a deterministic viral sequence retrieval tool (
gget virus) jumped from highly variable performance to near-100% accuracy on the VirBench dataset. 2 The chemistry paper moves in the opposite direction: it shows a general-purpose model, without bespoke tools, approaching dedicated-software accuracy on a domain-specific task through reasoning alone.Both are real results, and both tell the same story from different angles — the bottleneck in scientific AI is increasingly the quality of the surrounding infrastructure, not the model itself. When the infrastructure is good (deterministic retrieval tools for biology), cheap models with good tools beat expensive models without them. When the task is intrinsically reasoning-bound (spectrum interpretation), model capability still determines the ceiling.
Anthropic's implicit argument is that chemistry is moving into the second category faster than the field expected.
Añade más opiniones o contexto en torno a este contenido.